Number of coding ORFs
in the yeast genome
1. S. Cebrat, M.R. Dudek, A. Rogowska, 1997,
Asymmetry in nucleotide
composition of sense and antisense strands as a parameter for discriminating
open reading frames as protein coding sequences. Journal of Applied
Genetics,
38(1), 1 - 9. (abstract)
2. S. Cebrat, M. R. Dudek, P. Mackiewicz, M. Kowalczuk, M. Fita, 1997,
Asymmetry
of coding versus non-coding strand in coding sequences of
different genomes. Microbial & Comparative Genomics. 2(4),
259 - 268.
(abstract)
3. S. Cebrat, M. Dudek, P. Mackiewicz, 1997,
Is there any mystery
of ORPHANs? Journal of Applied Genetics,38(4), 365 - 372. (abstract)
4. S. Cebrat, P. Mackiewicz, M. R. Dudek, 1998,
The role of the genetic
code in generating new coding sequences inside existing genes. Biosystems,
45(2), 165 - 176. (abstract)
5. S. Cebrat, M. R. Dudek, P. Mackiewicz, 1998,
Sequence asymmetry
as a parameter indicating coding sequence in Saccharomyces cerevisiae genome.
Theory
in Biosciences, 117, 78 - 89. (abstract)
6. M. Kowalczuk, P. Mackiewicz, A. Gierlik, M. R. Dudek, S. Cebrat,
1999, Total Number of Coding Open Reading Frames in the Yest Genome.
YEAST,
15, 1031-1034. (abstract)
7. P. Mackiewicz, M. Kowalczuk, A. Gierlik, M. R. Dudek, S. Cebrat,
1999,
Origin and properties of noncoding ORFs in the yeast genome.Nucleic
Acids Res. 27(17), 3503-3509.(abstract)
8. P. Mackiewicz, M. Kowalczuk, D. Mackiewicz, A. Nowicka, M. Dudkiewicz,
A. Laszkiewicz, M. R. Dudek, S. Cebrat,
2002, How many protein-coding genes are there in the Saccharomyces
cerevisiae genome? Yeast 19(7), 619-629.
Protein coding ORFs have specific nucleotide
composition, which allows to distinguish between coding and noncoding sequences
(Cebrat et
al., 1997a, 1997b,
1998).
Compositionl trends in an ORF can be depicted by a DNA walk in two dimensional
space (see Graphic
representation of coding DNA sequences - a spider). To measure compositional
bias of protein coding sequences, we have calculated arcus tangent of (G-C)/(A-T)
for the first and the second codon positions of yeast ORFs. This parameter
measures the departure from A=T and G=C equimolarities within the
sense strand and allows us to present individual Open Reading Frames as
single points on the finite surface of a torus (see Distribution
of ORFs in a torus projection). Distribution of genes with described
phenotypes is much more compact than distribution of all ORFs. That means
that all genes that have been described so far have different nucleotide
composition than ORFs with unknown functions (Cebrat
et
al., 1998).
By comparing distribution of all ORFs
to distribution of the set of genes with known function, we have estimated
the total number of protein coding intronless ORFs longer than 100 codons
in the yeast genome at about 4700 (Cebrat
et
al., 1997a, 1998).
That estimation was confirmed for 4800 two years later using a larger pool
of known genes (Kowalczuk
et
al., 1999). This number should not be directly compared with the
estimations of the number of all coding sequences done by other authors
(which was done many times, leading to misunderstanding) because our number
has referred only to intronless ORFs longer than 100 codons and
not to all ORFs annotated in database (including intron-containing ORFs
and ORFs shorter than 100 codons).
In the paper (Mackiewicz et al.,
2002) we have approximated by two independent methods the total number
of protein coding sequences among all sequences annotated in MIPS (including
genes with introns and ORFs shorter than 100 codons as well) for 5300-5400.
Moreover, in this paper we have compared the results of estimations of
the total number of protein coding genes in the Saccharomyces cerevisiae
genome, which have been obtained by many laboratories since the yeast genome
sequence was published (MIPS database, Zhang and Wang, 2000, Souciet et
al., 2000, Blandin et al., 2000, Malpertuy et al., 2000; Wood
et al., 2001). It seems that the total number of protein coding
ORFs in this genome is several hundred lower than originally assumed (Goffeau
et al., 1996; Winzeler & Davis, 1997; Mewes et al., 1997).
However, it explains the "mystery
of orphans", which is the large number of ORFs with unknown function
and no homology to any known genes, discovered during systematic sequencing
of the yeast genome (Dujon, 1996). According to us, most orphans do not
code for proteins (Cebrat
et
al., 1997c). Nucleotide composition of many of them resembles the
antisense of genes, so they may have been generated by coding sequences
in the past and later moved to intergenic space by duplication (Mackiewicz
et
al., 1999, Gierlik
et
al., 1999). We have found about 700 ORFs in the MIPS database whose
putative protein products have homologues in frames different than the
frame which had been assumed in the database as coding (Mackiewicz
et
al., 1999).
Combined data of coding probabilities and compositional
parameters
counted by us for each ORFs and some data published by other authors are
available below.
Tabelarized
data:
Tha data for all chromosomes in Excel format: chr1_16.xls
chr1_16.zip
Tables description:
|
chr.
|
ORF's name
|
start
|
stop
|
length
|
S1
|
S2
|
V1
|
V2
|
D
|
cod. prob.
|
YZ score
|
MIPS
|
Genol.
|
T
|
Wood's annotation
|
brief ID
|
chr.
chromosome number;
length length
in codons; |
parameters of DNA asymmetry:
S1, S2
values of angles (in degrees) and V1,
V2
normalized length of vectors, for the first and the second codon
positions respectively;
D the
Euclidean distance of the ORF from the center of known genes distribution
in the four-dimensional space of parameters S1,
S2, V1, V2; |
| cod. prob. -
coding
probability (Mackiewicz et al.., 2002); http://smORFland.uni.wroc.pl; |
| YZ score
coding
probability according to Zhang and Wang (2000); |
MIPS class
in MIPS database (http://www.mips.biochem.mpg.de/proj/yeast/):
1-known protein;
2-strong similarity to known protein (higher than one third of FASTA
self-score);
3-similarity to known protein (lower than one third of FASTA self-score);
4-similar to unknown protein;
5-no similarity;
6-questionable ORF;
Similarities have been measured by FASTA scores. A FASTA score between
100 and 200 was defined as "weak similarity". FASTA scores between 200
and 1/3 of the selfscore (FASTA score of the protein, when aligned with
itself) of the protein were defined as "similarity". A FASTA score over
1/3 of selfscore was defined as "strong similarity". A questionable ORF
is defined by a combination of the following attributes: low CAI value,
partial overlap to a longer or known ORF, no similarity to other ORFs. |
Genol. class
in Genolevures program (Souciet
et al., 2000, Malpertuy et al.,
2000), http://cbi.labri.u-bordeaux.fr/Genolevures/Genolevures.php3:
0-probably spurious ORF;
1-ORF conserved in non-Ascomycetes;
2-ORF conserved in Ascomycetes only;
3-ORF without homology in orgs. other than S. cerevisiae itself;
T - total number of yeast species in which at least one homologue
of the S. cerevisiae ORF has been identified by Genolevures program; |
| Woods annotation re-annotations
of ORFs done by Wood et al. (2001); http://www.sanger.ac.uk/Projects/S_cerevisiae. |
Parameters describing asymmetry
of coding sequences:
To visualise asymmetry of DNA sequence and to show the
biological meaning of the parameters used, DNA walks are performed (Cebrat
et
al., 1997a, 1997b,
1998).
Imagine that a virtual walker starts its walk on the ORF sequence from
the first nucleotide of the start codon. Next, it jumps to the first nucleotide
in the second codon and so on (Fig.1A). It stops at
the first nucleotide of the last codon of the analysed ORF. These walks
(or jumps) are translated into a plot in a two-dimensional space where
the walker goes one unit up if the visited nucleotide is guanine, down
if the visited nucleotide is cytosine, right if adenine and left if thymine
(Fig. 1B). Then, the walker does its walk for the second
and the third codon positions. Since there are very strong and specific
compositional trends in each position in coding sequences, the plots drawn
by the walker are also specific for each position in codons and do not
resemble Brownian motion (Fig. 1C).
In this study, we have used two pairs of parameters describing
the obtained walks, which in fact are measures of the asymmetry in composition
of the first and the second positions in codons of ORF sequences:
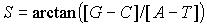 (eq. 1)
(eq. 1)
 (eq. 2)
(eq. 2)
where A, T, G, and C are the numbers of respective nucleotides
in the first or second positions in codons, and N is the length of the
analyzed ORF in codons.)
The parameter S (Fig. 1B) represents the angle between
x-axis and the vector determined by the beginning and the end of the corresponding
walk (eq. 1). It represents the relation between relative abundance of
purines over pyrimidines in complementary pairs of nucleotides. To avoid
infinite values of slopes, we have used a measure of angle rather than
tangent. Furthermore, the function of arctangent in many instances normalises
distributions. The parameter V (Fig. 1B) describing the asymmetry was the
length of the vector described by the beginning and the end of the walk
(eq. 2). The length of vector representing a sequence was divided by square
root of the length of this sequence in codons. Such normalisation shows
the relation between the length of this vector and the average vector for
random DNA sequence of the same length.
The parameter S allows us to present individual Open
Reading Frames as single points on the finite surface of a torus (see Distribution
of ORFs in a torus projection). In this case angle 1 corresponds to
X-coordinate and angle 2 to Y-coordinate. If you want to prepare ORFs distribution
of a chromosome, just plot the S1 values against S2 values. Then you will
find the position of any particular ORF in the distribution of all ORFs
of the chromosome.
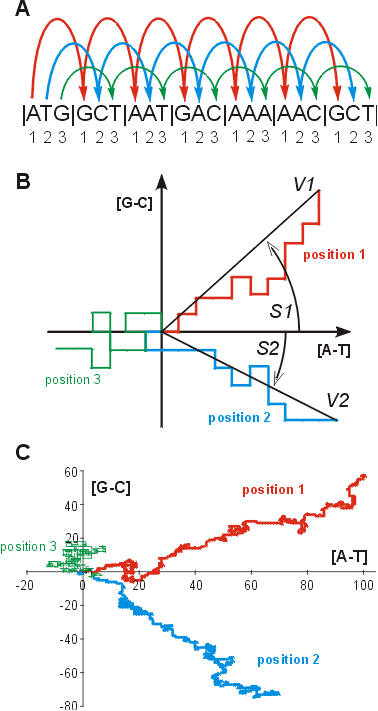 |
Figure 1. The method
of DNA walks on coding sequence:
(A) three DNA walks performed separately for each
position in codons beginning from start codon of ORF;
(B) two-dimensional representation of the above
three DNA walks. Two pairs of parameters describing asymmetry of the first
and the second positions in codons have been shown:
S - angle between a vector representing the walk and
x-axis,
V - length of this vector;
(C) two-dimensional representation of three DNA
walks performed for the gene SNF12 (YNR023w) coding component of SWI/SNF
global transcription activator complex. |
If you have any questions, do not hesitate to contact
cebrat@smorfland.uni.wroc.pl